Neuromyelitis Optica (NMO, NMOSD) Case Report Literature Review
Rodas Asrat, Sisay G, Ayub K, Semere N, Fethanegist T and DR Aduya
Rodas Asrat, Sisay G, Ayub K, Semere N, Fethanegist T and DR Aduya
St Paul’s Hospital Millennium Medical College, Ethiopia
*Corresponding author: Rodas Asrat, St Paul’s Hospital Millennium Medical College, Ethiopia, Tel: 215 926762207; Email: rodasasrat123@gmail.com
Received date: May 27, 2020; Accepted date: August 21, 2021; Published date: August 30, 2021
Citation: Rodas A (2021) Neuromyelitis Optica (NMO, NMOSD) Case Report Literature Review Stud IPJNCS Vol: 5 No: 4.
Abstract
A 47-year’s old female patient who is from Addis Ababa, Ethiopia which she was relatively healthy 7 years back Presented with decreased vision of her right eye, which progressed to complete visual loss. 3 months following the above symptom she started to experience decrease vision of left eye. Subsequently she presented with 4 days history of bilateral lower extremity weakness. Initially had paresthesia of lower extremities later lost sensation below her nipple line. To our knowledge, there is one case of NMO reported 3years back from our country, Ethiopia. Hence, we discuss the case presentation in light of the available literature on the topic. We will discuss some of the top differentials from list likely to most likely.
https://sporbahisleri.blogaaja.fi http://sporbahisleri.parsiblog.com https://spor-bahisleri.jimdosite.com https://sporbahisleri.edublogs.org https://sporbahisleri.websites.co.in https://sporbahisleri.podia.com https://sporbahisleri7.wordpress.com https://sporbahisleri.jigsy.com https://niwn-chroiaty-mcieung.yolasite.com https://spor-bahisleri.mywebselfsite.net https://sporbahisleri.mystrikingly.com https://sporbahisleri.splashthat.com https://sporbahisleri1.webnode.com.tr https://sporbahisleri.odoo.com http://sporbahisleri.creatorlink.net http://www.geocities.ws/sporbahisleri/ https://spor-s-site.thinkific.com https://artistecard.com/sporbahisleri https://sporbahisleri.estranky.cz https://spor-bahisleri.mozellosite.com https://651be6b563e56.site123.me https://betsitesiinceleme.blogspot.com https://sporbahisleri.hashnode.dev https://sporbahislerim.wixsite.com/spor-bahisleri https://sporbahislerix.weebly.com https://sites.google.com/view/betsiteleri https://codepen.io/sporbahisleri https://sporbahisleri.bcz.com https://www.smore.com/6rsb9
Keywords: Neuromyelitis optica; Multiple sclerosis, acute disseminated encephalomalitis (ADEM); Sub acute combined Nero degeneration, Neursarcoidosis; Ethiopia
Introduction
Neuromyelitis optica (NMO) is a rare condition, characterized by myelitis and optic neuritis, which shares a number of clinical and radiological features with multiple sclerosis (MS) [1–3]. The groundbreaking discovery of a novel, pathogenic autoantibody (termed NMO-IgG or AQP4-Ab) in a subset of patients by Dr Lennon and colleagues in 2004 [4, 5] has led to a tremendous increase in interest in NMO. NMO-IgG/AQP4 antibody-positive NMO is now considered a disease entity in its own right rather than a subtype of MS.
Neuromyelitis optica acuta‘ and the more rarely used English equivalent â??acute optic neuromyelitis‘ are both translations of the French term â??neuro-myélite optique aiguë‘, which was first used by Eugène Devic (1858–1930) in a paper communicated on the occasion of theCongrès Français de Médecine in Lyon in 1894. Devic intended the term to denote a novel syndrome characterized by acute myelitis and optic neuritis
In 2015, the International Panel for NMO Diagnosis proposed the unifying term of neuromyelitis optica spectrum disorders (NMOSD) for patients presenting selective demyelination of the spinal cord and the optic nerve. Specific criteria were established to facilitate earlier and more accurate diagnoses in AQP4 antibodies seropositive or seronegative patients presenting with ON, transverse myelitis, or area postrema clinical syndrome associated with a medullary MRI lesion.[6] Historically, NMO and MS are seen as overlapping central nervous system (CNS) inflammatory diseases. NMO was considered a variant of MS and the diagnosis of patients with NMO was delayed for many years due to the absence of specific criteria to recognize and distinguish the disease from MS.[7,8] With the discovery of AQP4 antibodies, the incidence of the disease started to be studied in a more accurate way [9,10].
In the following sections, we will provide a comprehensive review of NMO that covers its epidemiology, environmental risk factors, genetic factors, molecular mechanism, symptoms, comorbidities and clinical differentiation that we usually face difficulty while evaluating the patients, diagnosis, treatment, its management, and prognosis specific to our patient to our patient. In addition, we also provide a case discussion specific to NMO patients in our set up Ethiopia. There are few case reports from Africa [11, 12], perhaps because it is actually challenging to diagnose in resource limited settings.
Case report
History-A 47 year’s old female patient who is Christian in religion and married. Currently she came with a chief complaint of bilateral lower extremity weakness of 4 days duration. She was relatively healthy 7 years back at which time she presented with decreased vision of her right eye, which progressed to complete visual loss and dim vision of the left eye. It initially started from inability to see letters and progressed over a week to total vision loss of the right eye and after 3 months following the above symptom she started to experience decrease vision of left eye, which progressed to complete visual loss of the left eye. For this, the ophthalmology side was consulted and they wrote the following evaluation stating V=OD (NLP), OS(6/60, with pinhole 6/36) T=OD(37.2), OS(uncooperative).Optic atrophy, paleness and optic rim blurring (OD) and Pink disc (OS).So with the above evaluation they put the following Assessment=Optic atrophy (OD) 20 ? Optic neuritis 20?. So after they evaluated her they putted her on Prednisolone 60mg po/d (tapered over 2 months and the left eye vision improved).Then 4 years back, she presented to our ER with the complaint of abdominal pain of 5 days duration with failure to pass feces, flatus and urine. Up on physical examination Abdomen was tender, on PR hard and formed stool she was kept at emergency for 5 days and cleansing enema and catheterization was done with the Ass’t of Acute abdomen 20 ? Partial SBO 20 ? + AUR 20 ? .Since she was having lower back pain and radicular pain she was linked to AaBET Hospital for Neurosurgical intervention. There she was investigated with MRI with assessment of Ass’t : LBP with radiculopathy 20 ? DDD with moderate disc bulge at L5-S1 with spinal canal and left neural foraminal stenosis. She was given amitriptyline, tramadol, diclofenac and physiotherapy with little to no improvement. 4 months back with the same complaint she developed complete loss of vision of left eye and presented to optha clinic,PE=Optic atrophy, paleness and optic rim blurring (OD) and Slightly pale optic disc (OS). Ass’t - pupil involving CN III palsy + sudden loss of vision + Rt. Optic atrophy with optic neuritis. She was started on prednisolone 60mg/d. It was then tapered over the following one and half month with the return of her vision (near sight better than far sight).Currently she presented with bilateral lower limb weakness, which started over the right leg and progressed over 24hrs to involve the left leg. She also had tingling and burning sensation of the lower limbs initially which progressed to loss of sensation that progressed upward. She also has urinary and fecal retention. She is a known hypertensive patient for the past 18 years and on medication for the past 15 years on antihypertensive amlodipine 5mg/d po/d, HCT 25mg po/d, and methylopa 250 mg po/d. She is also a known dyslipidemic patient for the past 1 year on Atorvastatin 40mg po/d. She is adherent to her medications.
Personal History
Her meals usually contain of ‘enjera’ with ‘shiro’, vegetables, with occasional cooked meat. She is born and raised in Wolega and has 11 siblings (7 females and 4 males) who are all well and alive She is a para two mother, female and male, both are well and alive. She used to work as a janitor at Awash winery until 7 years back when she lost her right eye vision. Otherwise No Hx of Chronic cough, Eye pain, Diarrhea, nausea, vomiting, or hiccup, Nasal congestion, sneezing or rhinorrhea preceding the presentation, Joint pain, Easy fatigability, Skin rash or swelling over the neck or the axillae Other chronic illnesses (DM, asthma) Family history of the same illness
Physical Examination
G/A: conscious, alert and well communicative V/S:-BP=100-130/70-80 mmHg, PR=94-98 /min, RR=20-22 /min, SpO2 =94-98% on atm air LGS: no LAP INT/MSS: No rash PR: lax anal tone CNS: conscious, oriented to time place and person. Cranial nerves- CN ii= NLP on the Rt (consensual reflex intact), both intact on the Lt, CN iii=medial movement of the Rt is impaired and is slightly dilated. Lt has sluggish reaction to light and is midsize .Other cranial nerves are intact. GCS: 15/15 both short and long memory intact. Symmetrical muscle bulk, no fasciculation. Tone is hypotonic on both lower extremities and norm tonic on both upper extremity.
Reflex
| Upper Limb | RU | LU | Lower Limb | RL | LL |
|---|---|---|---|---|---|
| Biceps | 2/4 | 2/4 | Ankle | 1/4 | 1/4 |
| Triceps | 2/4 | 2/4 | Knee | 1/4 | 1/4 |
| Brachioradialis | 2/4 | 2/4 | Clonus (knee) | None | None |
| Babniski | equivocal | equivocal |
Power of the patient
| Upper Extremity | Right | Left | Lower Extremity | Right | left |
|---|---|---|---|---|---|
| Finger extension | 5/5 | 5/5 | Ankle Plantar flexion | 0/5 | 0/5 |
| Finger abduction | 1/5 | 2/5 | Ankle dorsiflexion | 0/5 | 0/5 |
| Finger flexion | 1/5 | 2/5 | Knee flexion | -- | -- |
| Wrist flexion | 5/5 | 3/5 | Knee extension | -- | -- |
| Wrist extension | 0/5 | 3/5 | Hip flexion | 0/5 | 0/5 |
| Elbow extension | 5/5 | 5/5 | Hip extension | 0/5 | 0/5 |
| Elbow flexion | 5/5 | 5/5 | Hip add. | 0/5 | 0/5 |
| Shoulder abd | 5/5 | 5/5 | Hip abd | 0/5 | 0/5 |
| Shoulder add | 5/5 | 5/5 |
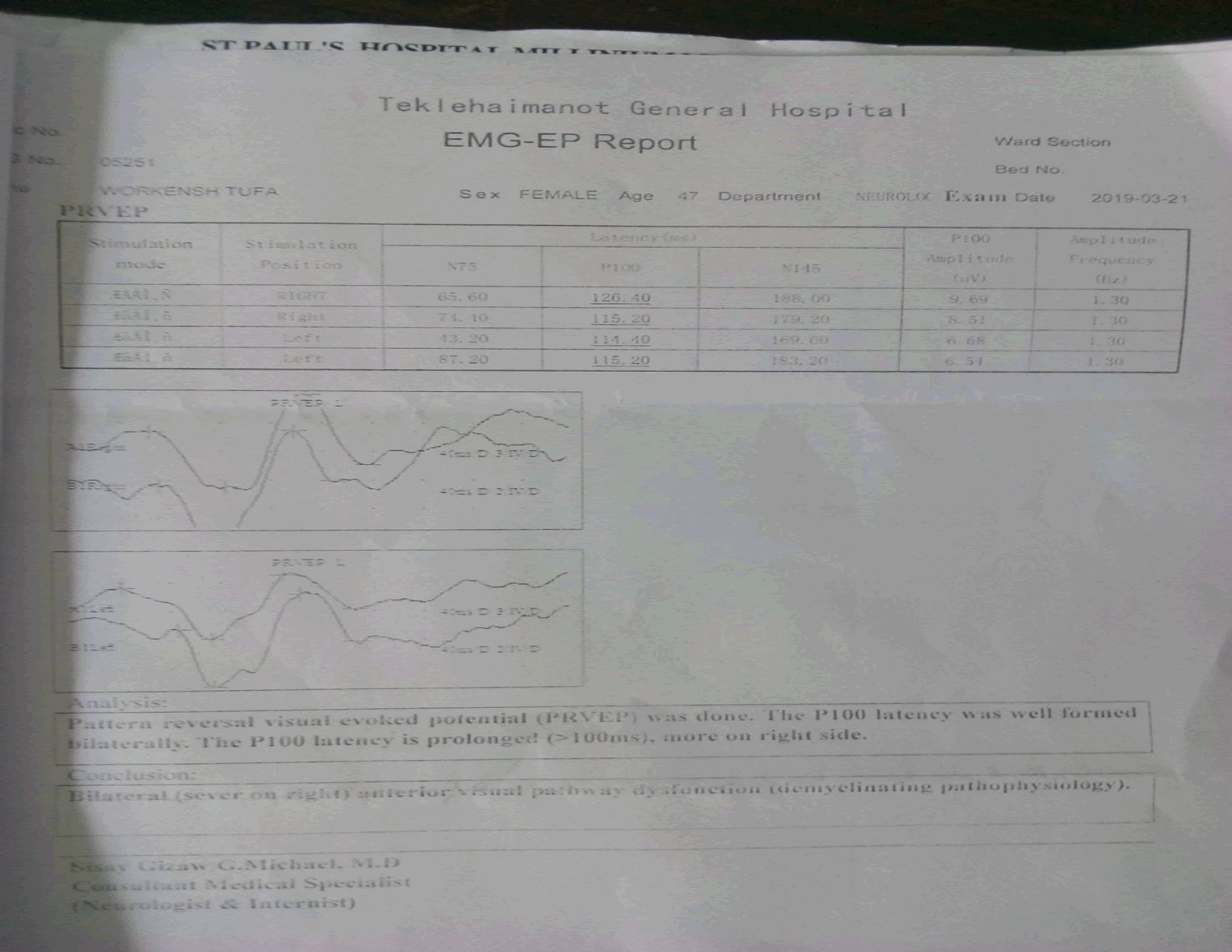
Sensory level is T4.Finger to nose test- normal .Rapid alternating movement – intact Investigation- Her laboratory investigations revealed normal complete blood count, liver function test, renal function test, serum electrolytes, and plasma glucose. She was also negative for syphilis (VDRL) and HIV tests. CSF Analysis-No cell, Glucose=82.4, Protein=24.8, Gram stain= negative. BRAIN MRI (Taken 7 YEARS BACK) = Normal. Brain MRI (taken 6 months back)= Prominent sulci and cortical volume loss at the left parieto-temporal lobe ,Few punctuate T2/FLAIR hyper intense lesions with in the deep white matter of the frontal and parietal lobes (Nonspecific white matter lesion),The orbital contents are unremarkable ,Left parieto-temporal volume loss secondary to ? Recent spinal MRI: Expansible long segment T1hypointense, T2 and STIR heterogeneously hyper intense lesion extending from C3 to lower thoracic vertebra. No contrast enhancement Echo- Mild concentric LVH with Grade I Diastolic Dysfunction with EF=60%Electromyography-Pattern reversal visual evoked potential (PRVEP) was done. The P100 latency was well formed bilaterally. The P100 latency is prolonged (>100ms), more on the right side. Bilateral (severe on Rt) anterior visual pathway dysfunction (demyelinating pathophysiology).
Discussion
Neuromyelitis optica (NMO) is characterized by optic neuritis and myelitis. NMO is distinct from classic relapsing-remitting MS with respect to pathogenesis, imaging features, biomarkers, neuropathology, and response to treatment. The target of the NMO antibody is the Aquaporin-4 (AQP4), a trans membrane protein which facilitates water transport in the CNS AQP4 is expressed in high levels within the optic nerves, hypothalamus, brain stem, periventricular, and gray matter of the spinal cord. Only 70% of NMO areAQP4-Ab-seeoposotive. 30% of patients with typical NMO are consistently seronegative for AQP4-antibody.Some of these patients present MOG-Abs. There have been evidences that MOG antibody is directly involved in the pathogenesis of MOG-antibody-associated disease. Prevalence ranges from 0.5 to 10 per 100,000.Incidence in women is up to 10 times higher over represented in some non-European populations worldwide, including Africans, East Asians, and Latin Americans.
Hallmark features of NMO include
Acute attacks of bilateral or rapidly sequential optic neuritis,with eye pain transverse myelitis characterized by symmetric paraparesis or quadriparesis, bladder dysfunction, and sensory loss below the level of the SC lesion paroxysmal tonic spams of the trunk or extremities, radicular pain, or Lhermitte sign
CNS involvement: intractable nausea, vomiting, hiccups, excessive daytime somnolence or narcolepsy, reversible posterior leukoencephalopathy syndrome, neuroendocrine disorders, and (in children) seizures 30–50% of NMO associate with other systemic or organ-specific autoimmunity, systemic lupus erythematosus,&Sjögren’s syndrome, autoimmune thyroid disease, myasthenia gravis, autoimmune-mediated vitamin B12 deficiency immune thrombocytopenic purpura, primary sclerosing cholangitis and ulcerative colitis
Cerebrospinal fluid-CSF restricted OCB, a diagnostic mainstay in MS, are present in only approximately 18% of AQP4- antibody-positive cases and frequently are transient.By contrast, OCB are present in far > 90% of cases in classical MS and detected over the entire course of the disease
CSF white cell counts (WCC) can be normal or mildly elevated in NMO (median 19/μl cells >100/μl are possible during relapse in 35% of cases. An elevated CSF protein 50% of cases, cytology often reveals, lymphocytic ,monocytic , neutrophilic and eosinophilic cell types which are usually absent in MS
MRI LETM onT2-weighted MRI, and primarily involving the central cord gray matter on axial sections, are highly suggestive of NMO. The cervical cord is affected in 60 % of cases, and lesions may extend into the medulla. At presentation, MRI of the brain is normal in 55 to 84 %. Overtime brain involvement develops in up to 85 % of patients.
Signs of poor prognosis
Frequent relapses during the first two years of disease
High severity of the first attack
Late disease onset
Coexistence of SLE or other non-organ-specific autoimmune disorders or the presence of autoantibodies
Diagnosis of NMO requires two absolute criteria, plus at least two or three supportive criteria. (1,6)
Absolute criteria:
- Optic neuritis
- Acute myelitis
Supportive criteria:
- Brain MRI not meeting criteria for MS at disease onset
- Spinal cord MRI with contiguous T2-weightedsignal abnormality extending over 3 or more vertebral segments, indicating a relatively large lesion in the spinal cord
- NMO-IgG seropositive status.
A case, which fulfills the above criteria, is 99% sensitive and 90% specific for Neuromyelitis optica.
Our patient was treated with Dexamethasone 1.5mg/kg started for 5 days followed by prednisolone 60mg/d for the next 7 day and followed by methylprednisolone1gm iv daily for five days and finally she was put again on prednisolone 60mg/d. Up on discharge she was put on azathioprine (1m/kg) which was 75 mg po/daily, Carbamazepine 200mg po BID, and predinisolone was tapered to 5mg po/day. Up on discharge She recovered some strength in her limbs with improvement of vision on the left eye but the right eye is still poor.. Her follow up after 7 months of treatment revealed that there is only some improvement in extremity strength; the muscle power had reached 3 out of 5 on the right lower limb and 2 out of 5 on the left lower limb with deep tendon reflexes, 1 out of 4 bilaterally. She has still constipation but the urinary incontinence has improved on follow up, where her last follow up was on March 17, 2020.Besides that her sensation is improved with no sensory level and gait disturbance .On her last follow up she was put on gabapentin 300mg po TID, Azathioprine 50 mg po/day, Amityptiline 25mg po/day.
In light of the above, the case presented here fulfilled the criteria for a diagnosis of NMO; the sole criteria, which does not met was a positive serostatus for NMO-IgG. The test was not done because it was not available in the country at the time of diagnosis
List of differentials to be ruled out (from most likely to least likely for this specific patient)
| DDX | In favor | Against |
|---|---|---|
| 1.NMOSD | 2 clinically evident lesions- Optic neuritis and myelitis-Female genderRelapsing remittingAgeEvoked potentialBlack raceLongitudinal extensive spinal cord lesionUMN lesion character with paraplegiaInitial brain MRIPresentation and duration | Absence of systemic auto immunities even if it is is no common (30-50%)Orbital contents were unremarkable on Brain MRI |
| 2.Multiple sclerosis | 2 clinically evident lesions- Optic neuritis and myelitis-Female genderRelapsing remittingAgeEvoked potential | Black raceBilateral ONAbsence of fatigueLongitudinal extensive spinal cord lesion |
| 3.Acute-disseminated encephalomalitis (ADEM ) | Bilateral ONTransverse myelitisLongitudinalExtensive hyperintense T2 weighted lesion | AgeNo preceding illness or vaccinationNo fever, change in mentation,- Not monophasicOnset of symptom not simultaneousCSF, no pleocytosis |
| 4.Sub acute combined neuro degeneration | MyelopathyOptic neuritis | Rapid onsetNo risk factor identifiedLETM |
| 5. Neuro sarcoidosis | Optic involvementMyelopathyBlack racefemaleAge | Rapid onset- Relapsing-Absence of involvement of other systems-Normal CSF protein- No cell in CSF-Normal chest X-ray |
Similar case was also,reported from our country, which is the only reported case from Ethiopian medical journal she was a 24-year-old female from Bedelle, Oromia region, Ethiopia who worked as a cashier. She presented to Tikur Anbessa Teaching Hospital on May 06, 2015 with a progressive quadriparesis involving the left arm, left leg, right arm, and right leg in that order, over a period of two weeks. She had lost her left eye vision progressively one week prior to the weakness. She also had associated tingling, numbness and burning sensation over her left upper limb. She had experienced similar attacks five years ago and another, just six months earlier, with fecal and urinary incontinence, which improved spontaneously over subsequent months. She was married and Nulliparous. Her past medical history showed no personal or family history of diabetes mellitus, hypertension, or cardiac illness. She did not have exposure to toxins or ever travelled out of Ethiopia. On physical examination, she was noted to have complete visual loss of the left eye with prominent retinal vessels and pale background on fundoscopic examination. All four limbs were flaccid with quadriparesis, with positive Babinski sign bilaterally. There was also sensory (touch, temperature and pain) loss of the left side of the body up to the mandible but there was no gait disturbance. The rest of the systemic examination was unremarkable. Her laboratory investigations revealed normal complete blood count, liver function test, renal function test, serum electrolytes, and plasma glucose. She was also negative for syphilis (VDRL) and HIV tests. Her brain CT scan and MRI findings were normal but cervical MRI (Figure1).revealed a hyper intense signal intensity (arrow) intramedullary lesion on T2W1 extending from C1-C7 vertebra. It was hypo intense on T1W1. The spinal cord was enlarged but the cervico-medullary junction was preserved. The MRI of the patient also showed a left side atrophied optic nerve.
Conclusion
Another case report from Uganda [11]. The case was a 24-year-old woman of Bantu origin who presented initially with bilateral loss of vision and weakness of the lower limbs in 2010 that resolved completely after a few days. Eight months later, she presented with bilateral lower limb weakness and urinary incontinence. Spinal MRI T2 weighted magnetic resonance imaging showed hyper intense ill-defined lesions from T4–T8, similar to our patient and the case from the USA. Her symptoms improved completely following steroid treatment. However, she developed an episode of quadriparesis four months later, which improved with steroids and azathioprine with some residual weakness requiring a walking aid.
From these cases, it is wise to consider NMO in patients (especially females) with unexplained recurrent quadriparesis and optic neuritis. Accordingly, spine MRI and serologic tests should be considered before starting patients on a specific treatment regimen.
References
- Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 9: 14.
- Wingerchuk DM, Hogancamp WF, Oâ??Brien PC, Weinshenker BG: The clinical course of neuromyelitis optica (Devicâ??s syndrome). Neurology 1999, 53:1107â??1114.
- Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, et al. (2012) Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 135: 1834â??1849.
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR: IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med2005, 202:473â??477.
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG: A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004, 364:2106â??2112.
- Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177â??89.
- Eaneff S, Wang V, Hanger M, Levy M, Mealy MA, Brandt AU, et al. Patient perspectives on neuromyelitis optica spectrum disorders: Data from the patientsLikeMe online community. Mult Scler Relat Disord. 2017;17:116â??22.
- Juryà ?czyk M, Craner M, Palace J. Overlapping CNS inflammatory diseases: Differentiating features of NMO and MS. J Neurol Neurosurg Psychiatry. 2015;86:20â??5
- Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet. 2004;364:2106â??12
- Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11: 535â??44.
- Kaddumukasa M, Musubire A, Kaddumukasa M, Matovu S, Katabira K. Neuromyelitis optica in a Ugandan woman. J Med Case Reports. 2013; 7:177.

Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences